Southern Blots
Technology
Blots
Gel electrophoresis provides the technology for separating nucleic acid fragments according to their size.
Gel electrophoresis relies on two characteristic properties of nucleic acids -the charge of the sugar-phosophate backbone and the uniform charge-mass ratio of all nucleic acids.(each base pair is associated with two negative charges)
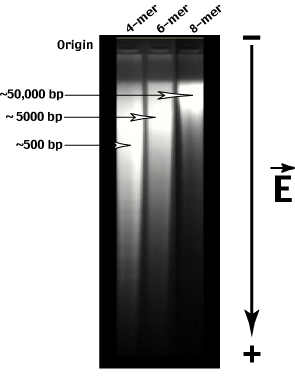
The gel matrix (typically agarose (large holes) or polyacrylamide (small holes)) acts as a sieve-like material with holes of uniform size. Small nucleic acid molecules can move through the gel quickly while large nucleic acid molecules move more slowly.
Notice that the genomic restriction digests do not generate a single size class of fragment. Instead the digest produces a 'random' smear of sizes from very large to very small. The AVERAGE size of the fragments produced can be calculated as on the previous page.
Gel electrophoresis allows us to fractionate genomic restriction fragments according to their length.
But how do we identify a specific fragment from all the others?
The identification of specific DNA sequences relies of complementary base pairing.
First, we need to denature the double-stranded DNA in the gel.
In general, two methods are available to dentature DNA or render it single-stranded. The first relies on heat - increased kinetic energy overcomes the H-bonds holding the double helix together and the two strands separate. Unfortunately, the gel material is heat sensitive - agarose melts at the same temperature as most ds-DNAs. We can also denature DNA chemically - exposure of DNA to high pH (0.2M NaOH) will also render it single-stranded without adversely affecting gel integrity.
A probe is a defined DNA sequence which will 'seek out' its complementary sequence in the population of genomic DNA fragments bound to the nylon filter. We will discuss hybridization a little later, but first we must find a way to label the probe sequence so that we can detect where on the size fractionated sample it has bound.
There are several types of labels we can incorporate into DNA probes. The simplest label is the incorporation of radioactive phosporus (32P) into the sugar phosphate backbone. This is easily done in a test tube (in vitro) by combining a DNA template (the probe), DNA polymerase, appropriate buffer and alpha-32P-dNTPs and allowing the polymerase to synthesize a new, labeled DNA strand. This radioactively labeled strand serves as our hybridization probe.
Other labels can also be incorporated into the probe sequence. The 5' phosphate can be replaced with a radioactive phosphate. Fluorescent tags can be incorporated by DNA polymerase. Other labels involving protein based detection systems can also be employed (digoxygenin-antibody detection, biotin-avidin detection etc etc etc.)
Non-specific binding of the probe to the filter and non-complementary genomic sequences are disrupted by reducing the salt and diluting the probe extensively - a process called 'washing'.
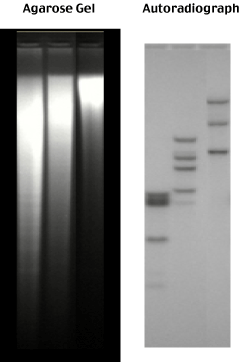
After washing off all the non-specifically bound probe, the blot is autoradiographed - a sheet of photographic film is placed on top of the blot.
Radioactive decay of the 32P in the probe produces a high energy beta particle (a high-energy electron) which exposes the siliver grains hit as the electron passes through the photographic emulsion.
The pattern of exposed silver grains in the film thus represents the decay of the probe specifically bound to its size fractionated complementary genomic sequence.
go to
Index